


Next: In more detail :
Up: A do-er's guide
Previous: One or more copies
Contents
- Prepare an ASCII file containing
h, k, l, Fo,
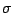 (Fo). Give it the name
data.hkl
(Fo). Give it the name
data.hkl
- Get a copy of the each of the PDB files containing the coordinates of
your search models. Edit the CRYST
cards, and give unit cell dimensions and space group number of the target
structure. Give them the names model1.pdb, model2.pdb, ...
NOTE WELL : You will need to have separate files for
each and every model present in the asymmetric unit of the target crystal
structure, even if these models correspond to the same molecule. To make
this clear : suppose that the target crystal structure contains two copies
of molecule A and two copies of molecule B. Then you need four PDB files :
model1.pdb and model2.pdb are identical and contain the
coordinates of molecule A. model3.pdb and model4.pdb are
identical and contain the coordinates of molecule B.
- Do Qs -reso <low> <high> -auto <xxx>, where <low> and <high>
are resolution limits, and <xxx> is the total number of
models (including multiple copies of the same molecule) that are present in the
asymmetric unit of the target structure (for the example above, Qs -reso 20 4 -auto 4).
Check that the
program goes through the initialisation phase and starts the actual minimisation.
- Start a proper batch job (and forget about it for a few days ? weeks ?).
Next: In more detail :
Up: A do-er's guide
Previous: One or more copies
Contents
NMG, January 2005